
Yes, we’re talking about FSHD
On those rare occasions (such as the FSHD Connect meeting) when large numbers of individuals with FSHD gather in one place, one can’t help but be struck by how greatly symptoms vary from person to person. Some twentysomethings are in wheelchairs, while some septuagenarians are still walking with the aid of a cane. One person’s face and body bear the unmistakable hallmarks of muscle loss. Another could pass for unaffected.
Such differences can be seen within a single family, even though all members have the same underlying FSHD genetics. Why so much variation? The answer may come down to a word much in vogue in biomedicine today: epigenetics.
In recent years, evidence of the essential role of epigenetics in FSHD has emerged from the laboratories of Michael Kyba at the University of Minnesota, Peter Jones at the University of Massachusetts Medical School, and an international collaboration led by Silvère van der Maarel, Rabi Tawil, and Stephen Tapscott. FSH Society grants supported the work in all three labs.
Classical genetics dealt with the inheritance of genetic traits encoded in the sequence of nucleotides that are strung together to form our DNA. Traits such as the hue of a pea flower (of Gregor Mendel fame) or an inherited disease such as sickle cell anemia are linked to the variations and misspellings of the genetic code.
But genes by themselves do not make a vegetable or animal. It is the expression of genes—the translation of the genetic code into proteins—that results in the final physical form and function of the organism. The human DNA—a six-foot-long chain—together with some RNA and protein, is packaged into a tightly coiled entity called chromatin, which forms the chromosomes inside every cell. The chromatin’s structure plays a key role in how the genes are expressed.
Epigenetics has to do with mechanisms other than DNA sequence that influence gene expression. One such mechanism is DNA methylation, which leads to changes in the conformation of the chromatin and, thus, the accessibility of the encoded gene. The more methylation, the tighter the chromatin is compacted and the less the gene inside is expressed. Conversely, reduced methylation (hypomethylation) relaxes the chromatin and increases the likelihood of gene expression.
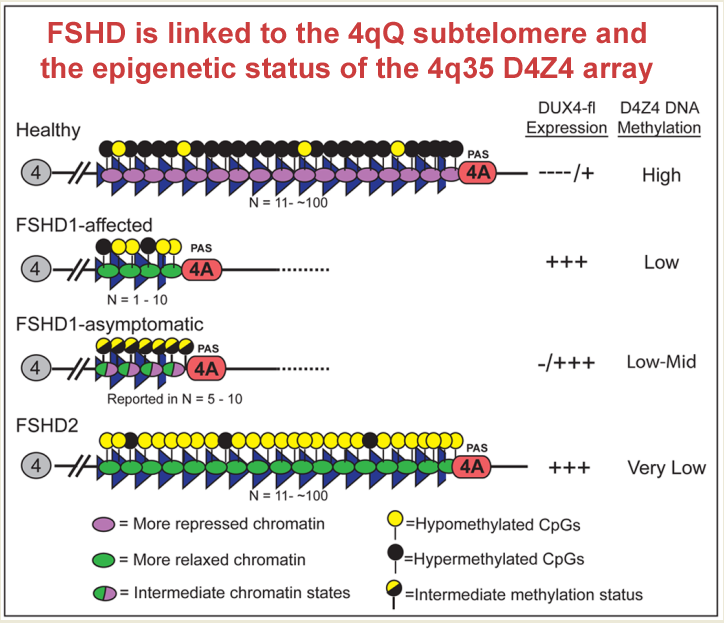
Enter FSHD. The most common form, FSHD1, is caused by a shortening of a region near the tip of the long arm of chromosome 4. This region consists of many repeating “D4Z4 units.” Normally, humans have between 11 to over 100 D4Z4 units in this location, but in individuals with FSHD1, there are only between one and 10 D4Z4 units. In addition, the region needs to be flanked by a “permissive” genetic structure called a PolyA signal in order for DUX4 to be expressed and for the disease to ensue.
The number of the D4Z4 units appears to correlate with how severe the disease is. Patients with between one and six units have more severe symptoms. But in individuals with seven to 10 units, the severity also depends on methylation: the less methylation (or more hypomethylation), the more severe the disease.
Between a person with full-blown FSHD and a relative with “non-manifesting” FSHD, the difference comes down to methylation. Both have the same number of D4Z4 units, but in the person with symptoms, the region is hypomethylated, whereas in the unaffected (non-manifesting) relative, the region is more methylated—not to the extent seen in healthy individuals, but at a level several times greater than seen in the relative with FSHD.
In FSHD Type 2 (FSHD2), individuals have normal numbers of D4Z4 repeats, but the entire D4Z4 repeat region is hypomethylated. FSHD2 is linked to mutations in the gene SMCHD1. Normal SMCHD1 plays a role in methylation of the D4Z4 repeat units, but the mutated gene results in less methylation, with some mutations being worse than others.
These findings are already leading to new diagnostic and treatment strategies. Exploiting the “epigenetic signature” of FSHD, the Jones group has developed a new laboratory test that can identify symptomatic FSHD1 and FSHD2, and clearly distinguish them from non-manifesting FSHD1 “carriers” and other types of muscular dystrophy. This new test “can be performed on genomic DNA isolated from blood, saliva, or cultured cells.” The Jones lab plans to develop its methods into an inexpensive new diagnostic test for FSHD.
In the Netherlands, a freshly launched biotech company, Facio Therapies, is taking aim at the SMCHD1 gene that causes FSHD2. Although FSHD2 comprises less than 5 percent of all FSHD cases, the company reasons that a drug that boosts SMCHD1 activity could elevate methylation of the D4Z4 repeat region in FSHD1 patients as well, repressing DUX4 expression and slowing the progression of their disease.
That, in any event, is the hope. Stay tuned. You are sure to hear much more about epigenetics in the years to come.
References
Hartweck LM, Anderson LJ, Lemmers RJ, Dandapat A, Toso EA, Dalton JC, Tawil R, Day JW, van der Maarel SM, Kyba M. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology. 2013 Jan 22;80(4):392-9. doi: 10.1212/WNL.0b013e31827f075c. Epub 2013 Jan 2. (PubMed)
Lemmers RJ, Goeman JJ, van der Vliet PJ, van Nieuwenhuizen MP, Balog J, Vos-Versteeg M, Camano P, Ramos Arroyo MA, Jerico I, Rogers MT, Miller DG, Upadhyaya M, Verschuuren JJ, Lopez de Munain Arregui A, van Engelen BG, Padberg GW, Sacconi S, Tawil R, Tapscott SJ, Bakker B, van der Maarel SM. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2015 Feb 1;24(3):659-69. doi: 10.1093/hmg/ddu486. Epub 2014 Sep 25. (PubMed)
Jones TI, Yan C, Sapp PC, McKenna-Yasek D, Kang PB, Quinn C, Salameh JS, King OD, Jones PL. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin Epigenetics. 2014 Oct 29;6(1):23. doi: 10.1186/1868-7083-6-23. (PubMed)
Himeda CL, Jones TI, Jones PL. FSHD as a model for epigenetic regulation and disease. AntioxRedox Sign. 2015 (PubMed)
From FSH Watch, Winter 2015
It is a pity a a terrible mistake that this brief article does not mention the key scientific contribution of Kowaljow et al 2007(1), a keystone publication that represents the foundation of the subsequent research on DUX4, proposed as the causative pathogenic protein in FSHD.
(1) The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Kowaljow et al.
Neuromuscul Disord. 2007 Aug;17(8):611-23. doi: 10.1016/j.nmd.2007.04.002.
I would like to thank you for this information. FSHD 2 is in my shoulders & my calfs. I am 75years old but I can walk with crutches. I have had both hips replaced.
Thank you
David Thomas.
M D UK No 12606